
Hi PAML Users,
Welcome to the PAML discussion group. This site is for posting questions and discussions of the PAML software package. PAML stands for Phylogenetic Analysis by Maximum Likelihood.
PAML是一个用最大似然法来对DNA和蛋白质序列进行系统发育分析的软件包,此软件由杨子恒教授开发,最新版本为v. 4.9 j。
此笔记主要用于记录PAML分析中遇到的一些问题,并长期更新。
参考链接
PAML主页
PAML手册
PAML FAQs
陈连福的博客
高芳銮老师的QQ空间
Evosite3D
Tutorial 2: Evaluating selection pressure with codon models
EVOLUTION AND GENOMICS
PAML discussion group
一. PAML可以做什么?
- 系统发育树的检验与比较;
- 复杂替代模型中参数的估计;
- 对不同假设模型进行似然率假设性检验;
- 基于全局或局部分子钟模型估算分歧时间;
- 使用核酸,氨基酸和密码子模型重构祖先序列;
- Monte Carlo模拟生成核酸,密码子和氨基酸序列数据集;
- 估算同义和非同义替代率和检测蛋白编码DNA序列中的正选择;
- 贝叶斯估算物种的分歧时间来合并化石尺度上的不确定性;
蛋白编码基因(protein coding sequence)的自然选择压力水平可以通过dN/dS(ω)值的大小来衡量,其中,dS代表同义替换率(synonymous rate),dN代表非同义替换率(non-synonymous rate)。在没有受到选择压力时,同义替换率和非同义替换率相等,此时dN/dS = 1;当受到负选择或净化选择压力时,自然选择会阻止氨基酸发生改变,同义替换率会大于非同义替换率,即dN/dS < 1;当受到正选择压力时,氨基酸的置换率会受自然选择的青睐,即蛋白功能可能会发生适应性改变,此时dN/dS > 1。
PAML简易流程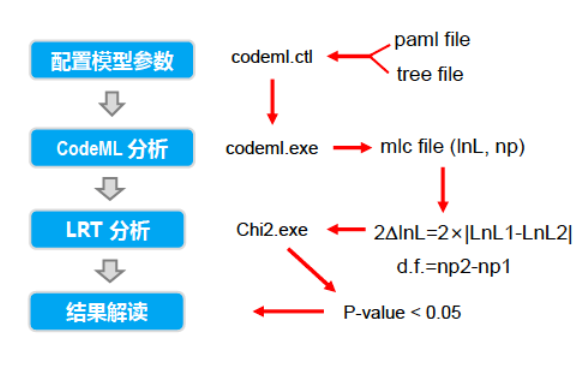
二. 如何运行PAML
要正常运行paml软件,需要四个标准文件:
- tree文件:Newick格式的树文件,主要包括分析涉及物种的系统发生关系,物种名必须与序列比对里面的物种名保持一致,否则会报错。
- fasta文件:fasta格式的多序列比对文件,通过要经过mafft、prank等软件进行比对,在经过gblocks、trimal等质量过滤软件处理后获得,序列长度必须为3的倍数且不允许存在终止密码子。
- codeml.ctl:paml软件的参数控制文件,通过修改该文件参数,实现模型切换
- codeml.exe:该软件的可执行文件
![]()

多序列比对与质量过滤在正选择分析以及系统发育研究中尤其重要,关乎到所有分析结果的正确与否。如何正确的处理编码序列,在之前的博文中已经详细说明,此处不在赘述。这里,主要强调一下控制文件的设置:1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39*输入输出参数:
seqfile = input.txt *设置输入的多序列比对文件路径。
treefile = input.trees *设置输入的树文件路径。
outfile = mlc *设置输出文件路径。
noisy = 9 *设置输出到屏幕上的信息量等级:0,1,2,3,9。
verbose = 0 *设置输出到结果文件中的信息量等级:0,精简模式结果(推荐);1,输出详细信息,包含碱基序列;2,输出更多信息。
getSE = 0 *设置是否计算并获得各参数的标准误:0,不需要;1,需要。
RateAncestor = 1 *设置是否计算序列中每个位点的替换率:0,不需要;1,需要。当设置成1时,会在结果文件rates中给出各个位点的碱基替换率;同时也会进行祖先序列的重构,在结果文件rst中有体现。
*数据使用说明参数:
runmode = 0 *设置程序获取进化树拓扑结构的方法:0,直接从输入文件中获取进化树拓扑结构(推荐);1,程序以输入文件中的多分叉树作为起始树,并使用星状分解法搜索最佳树;2,程序直接以星状树作为起始树,并使用星状分解法搜索最佳树;3,使用逐步添加法搜索最佳树;4,使用简约法获取起始树,再进行邻近分枝交换寻找最优树;5,以输入文件中的树作为起始树,再进行邻近分枝交换寻找最优树;-2,对密码子序列进行两两比较并使用ML方法计算DnDs,或对蛋白序列进行两两比较计算ML距离,而不进行其它参数(枝长和omega等)的计算。
* fix_blength = 0 *设置程序处理输入树文件中枝长数据的方法:0,忽略输入树文件中的枝长信息;-1,使用一个随机起始进行进行计算;1,以输入的枝长信息作为初始值进行ML迭代分析,此时需要注意输入的枝长信息是碱基替换率,而不是碱基替换数;2,不需要使用ML迭代计算枝长,直接使用输入的枝长信息,需要注意,若枝长信息和序列信息不吻合可能导致程序崩溃;3,让ML计算出的枝长和输入的枝长呈正比。
seqtype = 1 *设置输入的多序列比对数据的类型:1,密码子数据;2,氨基酸数据;3,输入数据虽然为密码子序列,但先转换为氨基酸序列后再进行分析。
CodonFreq = 2 *设置密码子频率的计算算法:0,表示除三种终止密码子频率为0,其余密码值频率全部为1/61;1,程序分别计算三个密码子位点的四种碱基频率,再算四种碱基频率在三个位点上的算术平均值,计算61种密码子频率时,使用该均值进行计算,这61种密码子频率之和不等于1,然后对其数据标准化,使其和为1,得到所有密码子的频率;2,程序分别计算三个密码子位点的四种碱基频率,计算61种密码子频率时,使用相应位点的碱基频率进行计算,这61种密码子频率之和不等于1,然后对其数据标准化,使其和为1,得到所有密码子的频率;3,直接使用观测到的各密码子的总的频数/所有密码值的总数,得到所有密码子的频率。一般选择第三种方法,设置CodonFre的值为2。第一种方法让所有密码子频率相等,不符合实际轻卡;第二种方法没有考虑到三种位点各碱基频率的差异,得到的密码子频率不准确;第三种方法能比较准确计算出各密码子的频率;第四种方法由输入数据得到真实的各密码子频率,但有时候有些非终止密码子的频率为0,可能不利于后续的计算。
cleandata = 1 *设置是否移除不明确字符(N、?、W、R和Y等)或含以后gap的列后再进行数据分析:0,不需要,但在序列两两比较的时候,还是会去除后进行比较;1,需要,**有可能会导致运行失败**。
* ndata = 1 *设置输入的多序列比对的数据个数。
clock = 0 *设置进化树中各分支上的变异速率是否一致,服从分子钟理论:0,变异速率不一致,不服从分子钟理论(当输入数据中物种相差较远时,各分支变异速率不一致);1,所有分枝具有相同的变异速率,全局上服从分子钟理论;2,进化树局部符和分子钟理论,程序认为除了指定的分支具有不同的进化速率,其它分枝都具有相同的进化速率,这要求输入的进化树信息中使用#来指定分支;3,对多基因数据进行联合分析。
Mgene = 0 *设置是否有多个基因的多序列比对信息输入,以及多各基因之间的参数是否一致:0,输入的多序列比对文件中仅包含一个基因时,或多个基因具有相同的Kappa和Pi参数;1,输入文件包含多个基因,这些基因之间是相互独立的(这些基因之间具有不同的Kappa和Pi值,且其进化树的枝长也不相关);2,输入文件包含多个基因,这些基因具有相同的Kappa值,不同的Pi值;3,输入文件包含多个基因,这些基因具有相同的Pi值,不同的Kappa值;4,输入文件包含多个基因,这些基因具有不同的Kappa值和不同的Pi值。当值是2、3或4时,多个基因的进化树枝长虽然长度不一样,但是呈正比关系。这点和参数值等于1是不一样的。
icode = 0 *设置遗传密码。其值1-10和NCBI的1-11遗传密码规则对应:0,表示通用的遗传密码。
Small_Diff = .5e-6 *设置一个很小的值,一般位于1e-8到1e-5之间。推荐检测设置不同的值在比较其结果,需要该参数值对结果没什么影响。
*位点替换模型参数:
model = 1 *若输入数据是密码子序列,该参数用于设置branch models,即进化树各分枝的omega值的分布:0,进化树上所有分枝的omega值一致;1,对每个分枝单独进行omega计算;2,设置多类omega值,根据树文件中对分枝的编号信息来确定类别,具有相同编号的分枝具有相同的omega值,没有编号的分枝具有相同的omega值,程序分别计算各编号和没有编号的omega值。
*若输入数据是蛋白序列,或数据是密码子序列且seqtype值是3时,该参数用于设置氨基酸替换模型:0,Poisson;1,氨基酸替换率和氨基酸的观测频率成正比;2,从aaRatefile参数指定的文件路径中读取氨基酸替换率信息,这些信息是根据经验获得,并记录到后缀为.dat的配置文件中。这些经验模型(Empirical Models)文件位于PAML软件安装目录中的dat目录下,例如,Dayhoff(dayhoff.dat)、WAG(wag.dat)、LG(lg.dat)、mtMAN(mtman.dat)和mtREV24(mtREV24.dat)等;
aaRatefile = dat/wag.dat *当对蛋白数据进行分析,且model = 2时,该参数生效,用于设置氨基酸替换模型。
aaDist = 0 *设置氨基酸之间的距离。
NSsites = 0 *输入数据时密码子序列时生效,用于设置site model,即序列各位点的omega值的分布:0,所有位点具有相同的omega值;1,各位点上的omega值小于1或等于1(服从中性进化neutral);2,各位点上的omega值小于1、等于1或大于1(选择性进化selection);3,discrete;4,freq;5:gamma;6,2gamma;7,beta;8,beta&w;9,beta&gamma;10,beta&gamma+1;11,beta&normal>1;12,0&2normal>1;13,3normal>0。
*可以一次输入多个模型进行计算并比较,其结果输出的rst文件中。
fix_alpha = 1 *序列中不同的位点具有不同的碱基替换率,服从discrete-gamma分布,该模型通过alpha(shape参数)和ncatG参数控制。该参数设置是否给定一个alpha值:0,使用ML方法对alpha值进行计算;1,使用下一个参数设置一个固定的alpha值。
*对于密码子序列,当NSsites参数值不为0或model不为0时,推荐设置fix_alpha = 1且alpha = 0,即不设置alpha值,认为位点间的变异速率一致,否则程序报错。若设置了alpha值,则程序认为不同密码子位点的变异速率不均匀,且同时所有位点的omega值一致,当然各分枝的omega值也会一致,这时要求NSsites和model参数值都设置为0(这一般不是我们需要的分析,它不能进行正选择分析了)。
alpha = 0 *设置一个固定的alpha值或初始alpha值(推荐设置为0.5)。该值小于1,表示只有少数热点位置的替换率较高;该值越小,表示位点替换率在各位点上越不均匀;若设置fix_alpha = 1且alpha = 0则表示所有位点的替换率是恒定一致的。
Malpha = 0 *当输入的多序列比对结果中有多基因时,设置这些基因间的alpha值是否相等:0,分别对每个基因单独计算alpha值;1,所有基因的alpha值保持一致。
ncatG = 8 *序列中不同位点的变异速率服从GAMMA分布的,ncatG是其一个参数,一般设置为5,4,8或10,且序列条数越多,该值设置越大。
*对于密码子序列,当NSites设置为3时,ncatG设置为3;当NSites设置为4时,ncatG设置为5;当NSites值设置>=5时,ncatG值设置为10。
fix_kappa = 0 *设置是否给定一个Kappa值:0,通过ML迭代来估算Kappa值;1,使用下一个参数设置一个固定的Kappa值。
kappa = 2 *设置一个固定的Kappa值,或一个初始的Kappa值。
fix_omega = 0 *设置是否给定一个omega值:0,通过ML迭代来估算Kappa值;1,使用下一个参数设置一个固定的omega值。
omega = .4 *设置一个固定的omega值,或一个初始的omega值。
method = 0 *设置评估枝长的ML迭代算法:0,使用PAML的老算法同时计算所有枝长,在clock = 0下有效;1,PAML新加入的算法,一次对一个枝长进行计算,该算法仅在clock参数值为1,2或3下工作。
树文件设置:1
2
3
4(((1,2),3),4,5);
((((1,2),3),4),5);
(((1:0.1, 2:0.2):0.8, 3:0.3):0.7, 4:0.4, 5:0.5);
(((Human:0.1, Chimpanzee:0.2):0.8, Gorilla:0.3):0.7,Orangutan:0.4, Gibbon:0.5);
需要注意的是,Newick格式的树尾部一定要有分号,没有的话程序无法正常运行。
三.PAML分析四大模型
在paml分析里面,主要涉及四种模型:位点模型(site-model)、支模型(branch model)、支位点模型(branch site model)以及进化支模型。位点模型通常适用于检测某一支系普遍性、广泛性的正选择,这种正选择是由于位点持续改变所引起的,例如,适应多种病原体;支模型主要是检测某一支系是否存在快速进化、选择压力约束以及正选择,但却无法检测到正选择位点;支位点模型比较准确与稳定,适用于检测某一支系断点性的正选择事件,此结果是由于适应某一时期环境改变所引起的,通常会保留在后代中;进化支模型(clade model),则主要是判别不同物种之间是否存受分化选择压力作用,不局限于正选择,它一次可以标记多个分支进行比较。
1. 位点模型
M0:假设所有位点具有相同的dN/dS值;
M1a:假设存在两类位点—保守位点0 < dN/dS < 1,中性进化位点dN/dS = 1,并且估算这两类位点的比率(p0,p1)和ω值(ω0,ω1);
M2a:假设存在三类位点——纯化选择位点dN/dS < 1,中性进化位点dN/dS = 1与正选择位点dN/dS > 1,并估算三类位点的比率(p0,p1,p2);
M3: 离散模型,假设所有位点的ω值呈离散分布;
M7:假设所有位点0 < ω <1且呈现beta分布;
M8:在M7模型的基础上,增加一类正选择位点(ω >1 );
M8a:与M8类似,只不过是将新增的ω固定为1;
位点模型的Codeml.ctl参数设置:1
2
3
4
5
6
7model = 0
NSsites = 0 1 2 3 7 8
即分别比较如下模型(* 为正选择模型,前提是需要LRTs显著):
M3(分散比率) vs. M0(负选择)
M2a* (正选择)vs. M1a (选择约束放松)
M8*(正选择) vs. M7 (选择约束放松)
M8* (正选择)vs. M8a(选择约束放松)
2.支模型
one rario:假设所有的进化谱系都具有相同的ω值;
free ratio:假设所有的支系都具有独立的ω值;
two ratio:假设前景支与背景支的ω不同
分支模型的Codeml.ctl参数设置:1
2
3
4
5
6
7two ratio: model = 2, NSsites = 0
one ratio: model = 0, NSsites = 0
free ratio: model = 1, NSsites = 0
one ratio(负选择) vs. free ratio(自由比率)
one ratio(负选择) vs. two ratio (正选择)
two ratio natural(中性进化) vs. two ratio (正选择)
3.支位点模型
假定位点间的ω值是变化的,同时也假定支系间的ω值是变化的。该模型主要用于检测前景支中正选择作用对部分位点的影响。
modelA null(零假设):ω值设定为固定值1
modelA(备择假设):估算其ω值是否大于1
背景支与前景支具有相同的位点ω值:
K0:前景支与背景支中的位点受到纯化选择0 < ω < 1;
K1:前景支与背景支中的位点处于中性进化0 < ω = 1;
背景支与前景支具有不同的位点ω值:
K2a:前景支处于中性进化,而背景支处于纯化选择;
K2b:前景支受到正选择压力( ω>1 ),而背景支处于中性进化;
支位点模型的的Codeml.ctl参数设置:
1 | ModelA: |
4.进化支模型
与枝位点模型类型,能同时检测多个进化枝(Clade),但是该模型并没有将背景支的dN/dS值约束在(0,1)。1
2
3
4M2a_model: model = 0 NSsites = 22
Clade Model C: model = 3 NSsites = 2
Clade Model D: model = 3 NSsites = 3
Clade Model C vs. M2a_model
四.PAML FAQ
1. 有根树与无根树
简单的区别方法就是,有根树的祖先节点是二叉树,而无根树为三叉树,举个例子:1
2
3
4
5((1,2),3); # 有根树
(1,2,3); #无根树
(((1,2),3),4); # 有根树
((1,2),3,4); #无根树
(1,2,(3,4)); #无根树
在使用codeml时,如果没有指定有根树参数却使用了有根树作为输入,那么在输出结果中将会得到这样的报错信息:"This is a rooted tree. Please check!"。对于大多数模型,即使使用有根树,其该模型似然值仍然是正确的,但是root周围两个分支的长度不稳定,因为它们的和是估计值。对于其他模型,似然估计和参数估计都是不正确的。因此,分析时确实应该注意到这一信息,并尽可能使用一棵无根树。我们可以使用R包ape将有根树转换为无根树:1
2
3
4
5
6
7setwd("C:/Users/lenovo/Desktop/")
library(ape)
tr <- read.tree("tree.txt")
unrooted_tr <- unroot(tr)
write.tree(unrooted_tr, "tree_unrooted.txt")
** 如果你有多个树需要转换,可以使用lapply函数操作**
write.tree(lapply(trees, unroot), "trees_unrooted.tr")
2.是否删除多序列比对中的gap与模糊字符
在多序列比对过程中,对齐gap是极其困难的,paml软件包没有办法无法处理gap。因此,我们可以通过设置cleandata = 1来去除gap;此外,还可以将gap当作为模糊字符进行处理。但是,这都不是最好的解决办法,这两种策略都低估了序列差异。就个人而言,我认为除了一个或两个序列之外,大多数序列都有序列信息的位点也许应该保留,而除了一个或两个序列之外,所有序列都有对齐间隙的位点最好被移除。因此,选择合适的多序列比对软件以及过滤软件,尤其重要。
3.当前景支的dN/dS值显著大于背景支时,该如何解释
如果检测前景支时,其dN/dS > 1时,我们可以认为它受到正选择作用。但是,如果其dN/dS <1但大于背景支时,就不能认为它是受正选择作用的,选择压力约束放松可能是较为合理的解释。此外,在Ohta's的微有害突变假说下,净化选择在大种群中比在小种群中更有效,因此不同谱系的种群规模的差异提供了另一个相容的假设。如果氨基酸的变化是稍微有害的,我们预计在大群体中它们从群体中移除的速度会比在小群体中更高。因此,即使两系在选择压力或基因功能上没有差异,我们也期望在一个大群体中看到一个较小的dN/dS比值,例如,许多核基因的dN/dS比值在啮齿类动物中低于灵长类或偶蹄类。
4.不同的模型鉴定出不同的位点,该选择相信哪一种模型?
通常,如果一个位点在一种模型下出现在列表中,那么在另一种模型下也会有相当大的概率。如果你以这种方式看待它们,结果可能没有太大不同。确定位点的问题很困难,而且容易出错。这种情况类似于从一个班级中得到排名前几的学生。列表包含的内容越多,质量就越差。因此,我们通常会认为后验概率大于95%或者99%的位点,是比较可信的。
5.如何标记前景支
如果你是关注某一个节点,则可以使用”#”;如果是关注某一类群,则可以使用”$”。对于位点模型以及free ratio分析,无需标注前景支;对于支模型以及进化支模型,单次分析可以标注多个前景支;但支位点模型单次分析仅能标注一个前景支。
1 | (((rabbit, rat) $1, human), goat_cow, marsupial) |
上面的两个例子其实是等同的,因此$1可以标记大的分支,包括其祖先节点以及现生节点。而#1仅仅是表示末端枝或者祖先节点。
这里有一些关于嵌套进化枝标签的规律。符号#的优先级高于\$,树顶端的进化枝标签优先于接近根处的祖先节点的进化枝标签。下面的两棵树也是一样。第一个树中\$1适用于整个胎盘哺乳动物的进化枝(除人类世系外),\$2适用于兔与鼠的进化枝。
1 | ((((rabbit, rat) $2, human #3), goat_cow) $1, marsupial); |
使用TreeView软件可以很方便的创建树文件并且检查树和标签是否正确。上面作为例子的树都可以被TreeView识别。在TreeView X中,你必须使用单引号来标记。如下:1
((((rabbit '#2', rat '#2') '#2', human '#3') '#1', goat_cow '#1') '#1', marsupial);
此外,还可以同时标注多个支为统一前景支,例如1
(((a #1, b) c #1 d) e)
6.基因复制后选择压力检测

某一基因,在动物祖先发生基因复制,分化为A,B两个不同支系。
7. dN/dS代表进化速率而非突变率
较高的dN/dS值可以解释为正选择或者快速进化。尽管突变本身也处于选择压力之中(大多数为净化选择),但不可以解释为“基因A增加了突变的选择压力”,因为突变是随机发生的。原则上讲,突变率会同时影响dN、dS,但通常dN/dS不受突变率影响。
dN/dS是一种进化速率,但它不是突变速率,因为同义和非同义替代率具有不同的选择约束水平。 选择压力测试的基本原理是:它假设同义替换是中性进行,也就是说,它们大多是在遗传漂移下进化的。如果这是真实情况,那么dS可以用作(中性)突变率的替代。然而,非同义替代率总是处于净化选择压力,在正选择下程度较小。因此,dN/dS是中性偏离的度量。所以,dN > dS,既dN/dS > 1,则受到正选择;如果dN小于dS,则dN/dS < 1,则是净化选择。选择压力测试的关键就是它通过特定基因的同义替换的“中性”进化速率归一化非同义替换的速率。
8.基因树or物种树
在任何情况下,使用代表真实演化历史的基因树都是最好的。但是,有时可能无法轻易判断是否符合真实演化历史,那么你可以选择物种树作为代替。基因组水平分析,那么推荐使用物种树。你可以可以使用基因树和物种树进行数据稳健性测试。
9. 祖先节点有正选择信号而整个枝系却没有检测到如何解释
如果以哺乳动物祖先为前景,假设该基因在共同祖先中存在适应性,可能是由于获得了新的功能,但随后该基因在净化选择下得到了保守进化。如果你把整个clade作为前景,那么假设该基因在整个哺乳动物中的所有分支都承受着持续的压力来改变或多样化,如果该基因涉及到防御或免疫,则可能是这种情况。
对于到底是检测祖先,还是整个枝系,取决于生物学问题。例如,溶菌酶在所有colobine猴子中都应该具有相同的功能,因此蛋白质在clade内被期望受到选择性约束,但在colobine clade的分支上,该酶显然获得了一个新的功能,其正选择驱动了氨基酸的变化。有了这个假设,你就应该把分支的祖先贴上clade的标签,而不是那些在clade中的分支。
10.Clade模型判别背景支是否同样受正选择
经常遇到审稿人的审稿意见是这样的:前景分支上正选择的基因的重要支持并不意味着在背景分支上没有正选择,这些基因可能仍在许多(即使不是全部)背景分支上处于正选择状态。为了检验原假设(基因仅在前景支受到正选择)是否正确,可以进一步通过Clade模型检验,因为进化枝模型允许在不限制背景dN/dS小于1的约束下估计前景支与背景支dS/dN的比率。
11.基因受到正选择但无正选择位点
一种可能性是,对整个基因有积极选择的证据,但每个单独位点的信息或证据太弱。 可以查看rst文件,该文件具有所有站点的后验概率,以查看是否是这种情况,mlc文件只列出后验概率高于0.5的文件。
12. 有正选择位点但是通过序列比对却找不到
可能是因为codeml在删除gap或模糊字符的列,之后重新编号位点了(cleandata =1)。
13. dN/dS(ω)值过大,结果是否可信?
遇到这种极大值的dN/dS时,比如ω = 999,首先要确保你的序列是否正确;其次,该位置的dn,ds是否远小于0.0001,枝长是否过小。显然,高度相似的序列和非常发散的序列都不是信息丰富的,很难指定确切的值。为了避免此类问题的出现,可以先通过M0模型获得枝长,再将具有分支长度的进化树应用到codeml,并在ctl中设置FIX_blength=2。
14. Branch-site检测适应性趋同进化

如图所示,红色分支代表表型趋同的进化分支,如果你想通过分支位点模型来检测适应性趋同进化,应该将所有红色分支统一设置为前景分支。当然,前提是假设所有前景支均具有相同的位点受到正选择。 对于背景支是否同样存在类似的适应性趋同,可以通过clade进化支模型进行检测。
15. 若p2(正选择位点概率)接近于0
p0/ω0,p1/ω1,p2=(1-p0-p1)/ω2:在正选择分析的备择假设结果文件中,通常会获得三个p值,其中,p0代表处于净化选择下的位点概率;p1代表处于中性进化下的位点概率;p2则代表处于正选择下的位点概率。
16.分支模型检测正选择)
可以使用two ratio备择假设(fix_omega = 0 omega = 1)与two ratio零假设(fix_omega = 1 omega = 1)进行统计假设检验。
17. 选择约束放松
codeml检测选择约束放松可以通过2步完成:首先,确定dN/dS显著增加的情况(由于正选择或者选择约束放松);然后,过滤掉显著正选择的情况。
18. 不同模型下的起始ω
针对于CladeC和CladeD模型,一般要设置几个不同的起始ω,测试其lnL值是否稳定(ω=0.001,ω=0.01,ω=0.1,ω=0.5,ω=1,ω=1.5),最终一般会选取lnL较大的值作为最终值。
19. 枝长会影响PAML结果
正常分析,应该先用M0估计树的枝长,Kappa值,然后用跑出来的树作为初始树,并设置fix_blength = 2。
20. 基因家族选择压力分析
21. CladeC检测正选择
CladeC经常用于检测,不同分支的分化选择压力,但有时候检测到前景枝dN/dS>1,此时我们需要进一步利用CladeC的零假设进一步测试(fix_omega=1,omega=1),或者利用branch-site进一步验证一致性。
22. Free ratio检测结果是否可以作为正选择的证据
free ratio模型估计通常会造成较大的抽样误差,例如,较短的分支通常会具有较大的dS/dN。所以,一般对于free ratio出现dN/dS > 999或者dN、dS < 0.0005结果不建议采用。
23. dS>5
24. CladeC模型检测多组支系选择压力差异
在dataset中,有三个clade:A, B, C,那么在clade 1和clade 2之间是否存在显著差异?
假设,前景枝的标注如下所示:1
((A1,A2)$1 , (B1,B2)$2 , (C1,C2)$0);
首先,为了测试clades之间的显着差异,您可以比较CladeC与M2a_rel。M2a_rel假设$1、$2、$0等都是在相同的选择压力下进化的,这个测试应该有两个自由度。1
(A1,a2)$1,(b1,b2)$1,(c1,c2)$0);
其次,为了测试clade A和B之间的显着差异,同时允许clade C是不同的,您可以比较使用上面提供的树运行CMC与使用更简单的树运行CMC的契合程度。在这种情况下,更简单的树会将clade A和B分配给同一个组,这个测试应该有一个自由度。如下所示:
25. 多个前景支的支位点模型检测
当数据集中有多个前景支时:1)进行多个测试,然后在每个测试中设置一个感兴趣的分支作为前景分支;2)只进行一次测试,在此测试中中将所有感兴趣的分支设置为前景分支。那么,此时又会出现另一个问题就是进行多个测试时其他感兴趣分支是否应该被去除?这也许应该取决于具体的生物学问题。

