前几天,实验室的师弟师妹通过本地blast获取一些没有基因组注释物种的蛋白编码序列。原本以为可以快速地进行下一步的选择压力分析,没想到却在多序列比对这一环节出现了棘手的问题。以前,我都是经过PRANK软件进行多序列比对,然后再使用Gblocks软件对数据进行过滤的。现在,由于师弟师妹在拼接CDS序列时,有些碱基并不是保留3的倍数,造成很多编码序列出现移码突变,甚至变成了伪假基因(几百个基因)。虽然,现在可以进行多序列比对的软件有很多,比如Muscle、MAFFT、PRANK等,但它们均不能解决移码突变的问题。于是,我开始去google搜寻解决方案。功夫不负有心人,我最终找到了完美的解决办法,收获颇丰,所以把这个过程记录下来。
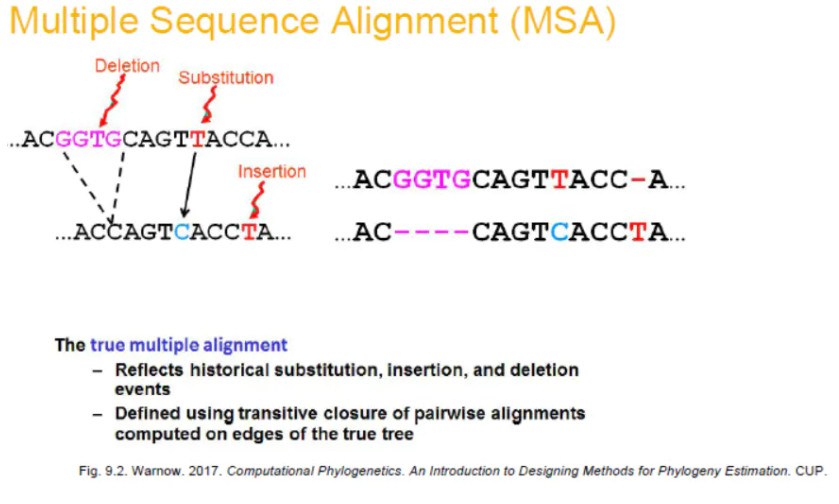
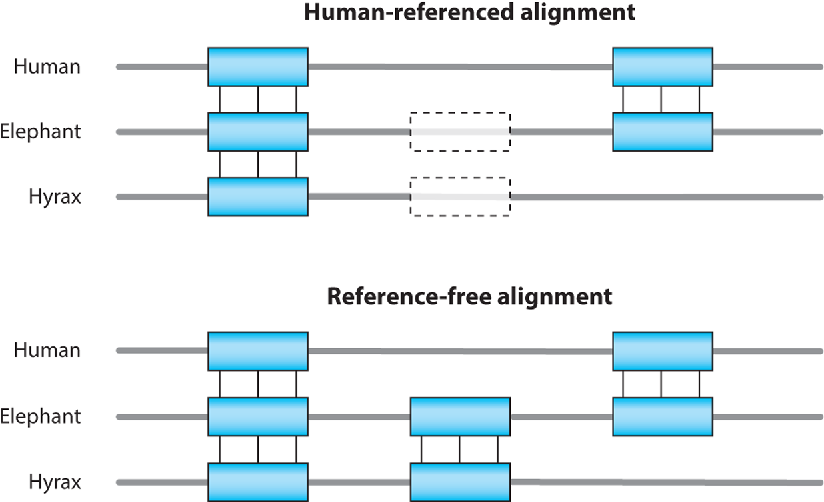
多序列比对(multiple sequences alignment,MSA)是开展进化生物学研究的前提,后续可以进行选择压力分析以及推断物种系统发育历史。
多序列比对表示不同序列中的氨基酸/核苷酸的位点同源性。将序列比对用于进化分析时,处于相同位置的氨基酸/核苷酸位点则被认为在进化上是同源的,并且具有共同的祖先。通过研究序列比对中同源序列是如何随时间变化的,可以推断序列的结构和功能是如何进化的。比如,对多个物种同源序列的比较可以发现发生替换的位点,进而判断出哪个序列在哪个位置发生变化。另外,两个序列之间的长度差异可以通过删除一个序列中的现有碱基位点或插入另一个序列中的新碱基位点来解释。两个序列之间发生替换差异的进化谱系,可以用外群序列来解决,也可以通过推断两个序列的祖先的特征状态来解决。因此,以上所有情况的结论准确性很可能取决于多序列比对(MSA)的准确性。
最近的研究表明,MSA算法在分析基因组序列时会产生不同的结果,包括系统发生树推断和适应性进化的检测。换句话说,错误的MSA将会产生一个非真实的进化历史信号,从而导致错误的推断。为了减少MSA错误的影响,许多研究人员在优化MSA算法和MSA质量过滤软件等方面做出了大量的努力。然而,MSA不仅包含比对错误,而且还包含序列自身错误(如测序错误、组装错误或错误的基因注释等)。所以,正确选择MSA分析流程可以显著减少这些错误的产生,从而获得高质量的保守同源序列用于下游的分子进化分析。
接下来,我主要是介绍一些平时比较常用的主流分析软件以及它们的优缺点。可能还有很多优秀的软件,由于个人精力有限,暂时只讲一下我自己比较熟悉的工具,请大家见谅。
01. Multiple Sequence Alignmen
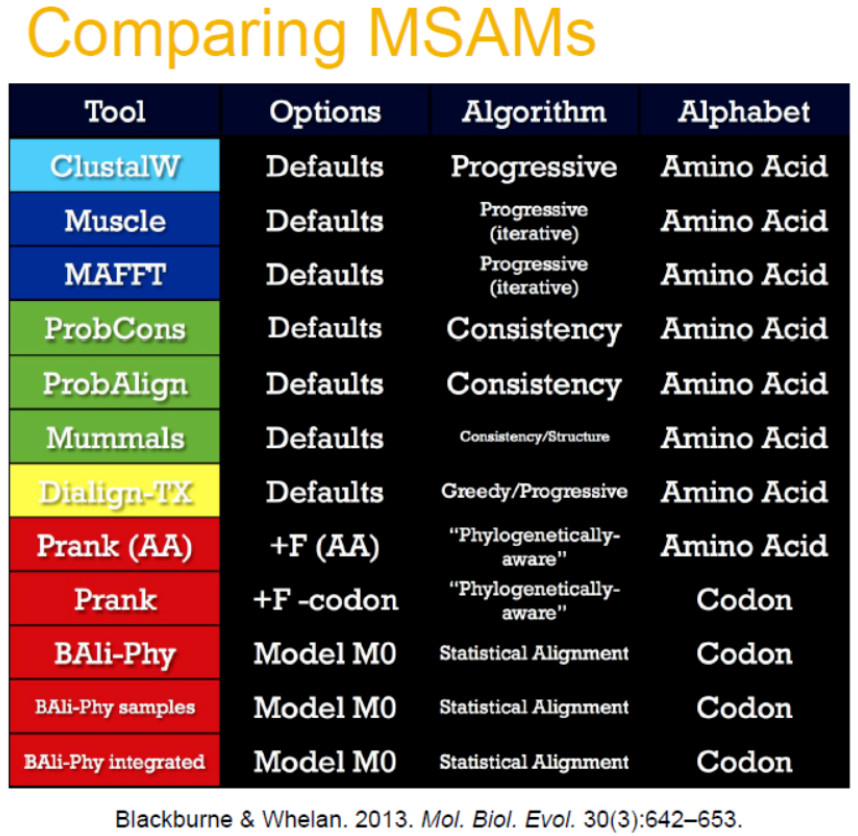
2013年,Blackburne等人在《Molecular Biology and Evolution》期刊发表他们的研究成果,该成果证明了MSA方法在下游分析中的关键作用,突出了在分析中选择的不同的MSA软件对其结果有明显的影响。
Clustal是最早开始使用的多序列比对工具,由 Feng 和 Doolittle 于1987年提出,该程序有许多版本,可以基于多种平台,目前引用次数已近100000次(Thompson et al. 1994; Larkin et al. 2007)。它采用一种渐进的比对方法,先将多个序列两两比对构建距离矩阵,反应序列之间两两关系;然后根据距离矩阵计算产生系统进化指导树,对关系密切的序列进行加权,然后从最紧密的两条序列开始,逐步引入临近的序列并不断重新构建比对,直到所有序列都被加入为止。因此,它是一种试探算法,所以渐进比对不能保证能够得到最优的比对。
Muscle是一款速度最快的比对软件之一,在速度和精度上都优于ClustalW,目前引用次数高达26246次(Edgar 2004)。Muscle采用的是迭代方法进行比对运算,每一次最优化过程就是迭代过程,通过不断地使用动态规划法重排来纠正这种错误,同时对这些亚类群进行比对以获得所有序列的全局比对。一个形象的例子是,10000条长度为350bp的数据进行比对只需要十几分钟,而用ClustalW则可能需要1年。但是,速度快的后果就是准确度降低。
MAFFT的第一个版本于2002年发布,它使用了一种基于渐进对齐的算法,利用快速傅里叶变换对序列进行聚类(Katoh K et al. 2002)。其后版本的MAFFT增加了其他算法和操作模式,包括更快地对大量序列进行比对的选项、更高精度的比对、非编码RNA序列的比对以及在现有比对中添加新的序列,目前其引用次数也近达20000次。它的比对精度要高于Muscle,速度也较快,但相比其他依赖物种进化关系的比对软件,它的准确度还有一些差距。
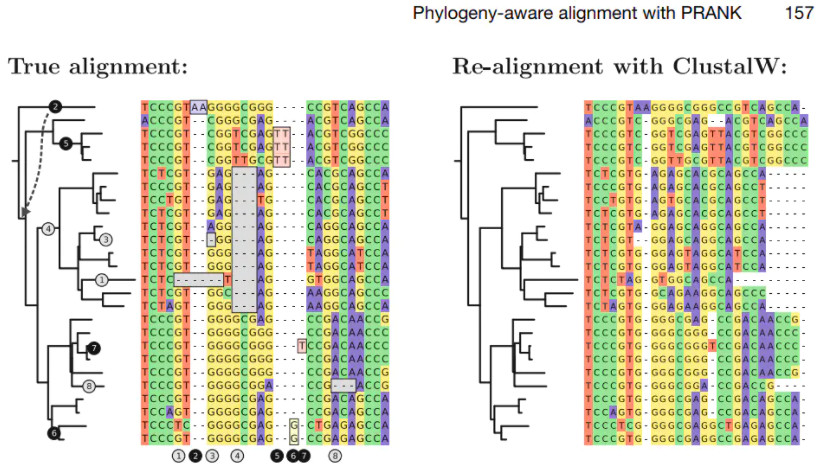
PRANK是由 Löytynoja 于2005年开发的软件,目前它的引用数达到1662次。它是一种针对DNA、密码子(codon)和氨基酸序列的概率多重比对程序,基于一种新的算法,能够确定各序列在与其共同的祖先分化后发生的碱基替换,插入(insertion)或缺失(deletion)事件。此外,PRANK可以重新构建其祖先序列,有DNA翻译以及回译选项( DNA translation/back-translation)。最后,由于PRANK比对准确度高,相对比较耗时,不太适合基因组数据分析。
BAli-Phy是一种贝叶斯后验比对软件,它利用马尔可夫链蒙特卡罗来探索给定分子序列数据的比对和系统发育的联合空间,同时估计消除了对不准确的比对引导树的偏差,在比对过程中采用了更复杂的替换模型,并且自动利用共享插入/删除中的信息来帮助推断系统发育关系(Suchard et al. 2006)。其准确度,与PRANK几乎差不多(我自己并没有使用过这款软件)。
Blackburne 的研究结果显示,基于进化方法的PRANK与BAli-Phy软件,可以根据序列的差异性灵活地使用不同的比对标准(评分矩阵与罚分),然而ClustalW、Muscle以及MAFFT等经典比对软件则不能。他们的结果表明,在构建序列比对时把物种的进化关系及距离考虑进去能有效提高比对准确性,因此比起仅应用新的计算方法,可能会产生更大的改进潜力。
除此以外,还有许多研究也同样揭示ClustalW、Muscle以及MAFFT等经典比对软件在MSA精确度方面还存在许多缺陷(如下图所示)。
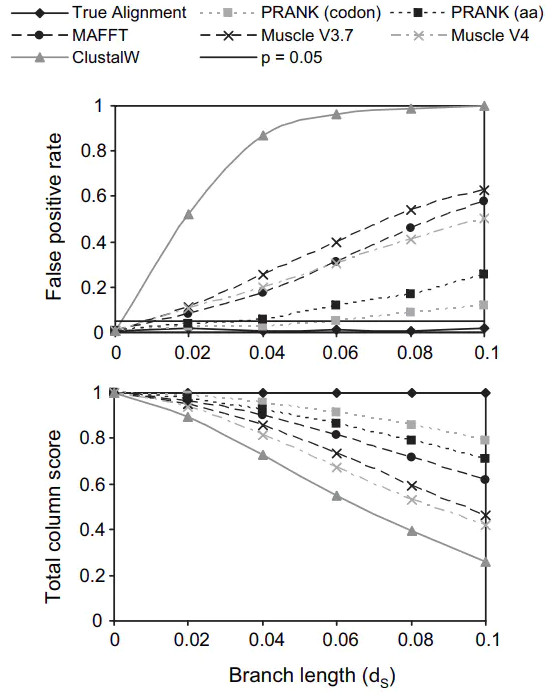
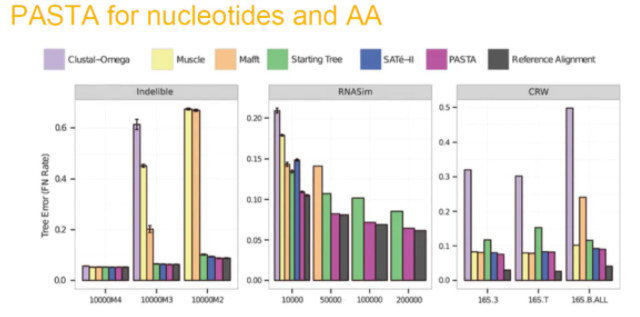
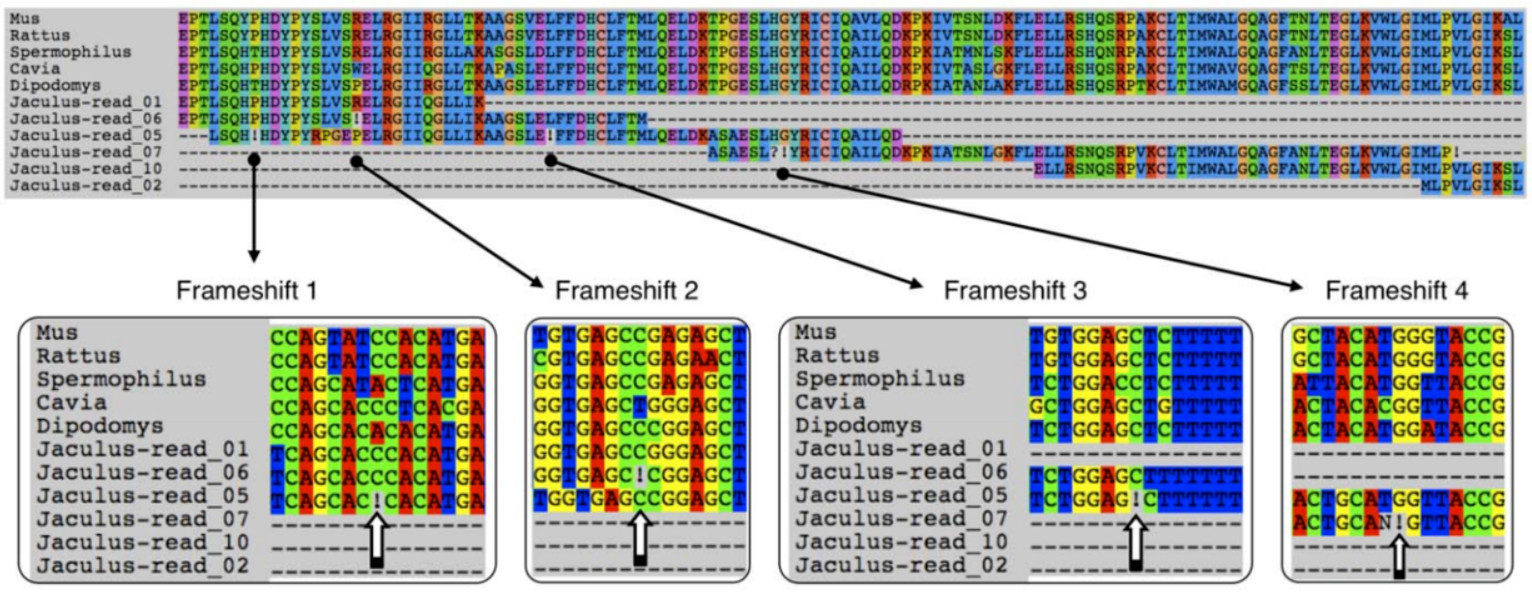
MACSE是第一个可以用于自动调整含有移码变异以及假基因的蛋白编码序列,而不破坏潜在密码子结构的多序列比对工具(Ranwez et al. 2011)。它的关键特征是在核苷酸水平上对DNA序列进行比对,但有可能包括不是三个碱基的倍数的间隙长度,即产生移码,同时基于其氨基酸翻译对产生的核苷酸比对进行评分。这使得可以产生保留潜在密码子结构的核苷酸比对,同时受益于氨基酸序列的更高相似性(Ranwez et al. 2018)。现在,这个软件已被许多基因组分析所广泛使用,其准确度也相对较好,然而目前并没有研究去仔细分析过它的性能。
OrthoMAM v8/v10数据库就是采用这个软件的分析流程:CDS and exon sequences are aligned at the codon level in two steps. First, the translated amino acids are aligned using MAFFT (Katoh et al. 2005) and gaps are reported onto the nucleotide sequences. This alignment is then refined using MACSE (Ranwez et al. 2011) to obtain a final codon alignment unaffected by frameshifts, misassemblies, and sequencing errors. Amino acid alignments are then filtered to reduce the impact of errors on evolutionary inferences using HMMcleaner (Philippe et al. 2017).
此外,还有几种基于隐马尔科夫模型(Profile HMM Methods)的多序列比对工具,这些工具目前我还不清楚它们的具体优缺点。
- SEPP(SATé-enabled Phylogenetic Placement):解决将short reads放入参考序列和树的系统发育问题;
- TIPP(Taxonomic Identification and Phylogenetic Profiling):解决元组数据的分类识别和丰度分析问题
- UPP(Ultra-large alignments using Phylogeny-aware Profiles):解决非常大的数据集对齐的问题,这些数据集可能包含一些零碎的数据,可以将数据集多达1,000,000条序列对齐;
- HIPPI(Highly Accurate Protein Family Classification with Ensembles of HMMs):解决蛋白质家族分类的问题;
02. Trimming

当获得精准的多序列比对后,接下来的任务就是要过滤掉一些低质量以及高变异度的序列区域,仅保留进化保守的区域用于后续分析。其中,产生这些低质量区域的原因主要有两个方面:
生物学因素:如果比较来自远缘物种的蛋白序列,很有可能只有蛋白质的功能部分在序列上具有较高的保守性。其他部分,如第一个外显子或最后一个外显子区域,更有可能改变它们的氨基酸序列,无论是在氨基酸含量方面,还是在insertion/deletion存在的情况下。即使当多个序列对齐的预测是正确的,它也可能对系统发育树的推断产生负面影响;
错误:测序错误,基因组组装错误、基因预测错误以及多序列比对错误;
目前主流的多序列比对过滤软件可分为以下几个方面:一是,block-filtering:trimAl、Gblocks;二是,segment filtering:HmmCleaner、PREQUA;三是,sliding window analysis:FasParser2。
Gblocks 是较早时间(2000年)就应用于MSA过滤的软件,由Castresana实验室所开发,被引用次数为6194次。它可以将MSA中大片断非保守性或者是非同源片段给删除(6-10bp左右的非同源片段则不能很好识别出来),另外它还对Block(即一段连续的且中间不含Gap的列)的长度进行了限制。有研究把这种方法应用于构建系统发育树,发现通过删除原始比对中得分比较低的片段以后,Gblocks有效地提高了系统发育关系的确定性(Talavera et al. 2007)。但是,Gblocks的问题在于它太武断地规定了某个具体的阈值来判断比对片段的保留或删除。比较科学的阈值选择应该要根据序列的具体情况来确定,而不是对所有基因的比对都采用同一个阈值,因为不同的基因或同一个基因的不同片段,其进化速率可能都不相同。
trimAl Capella-Gutiérrez等人于2009年开发的另一款MSA自动调整过滤软件,特别适用于大规模的系统发育分析,目前引用次数已有2157次。它的主要优点就是速度快,准确度高,相比于Gblocks,它可以自动选择在每个特定比对中使用的参数,主要包括Gap的比例以及氨基酸相似性的水平,从而优化 signal-to-noise ratio 。
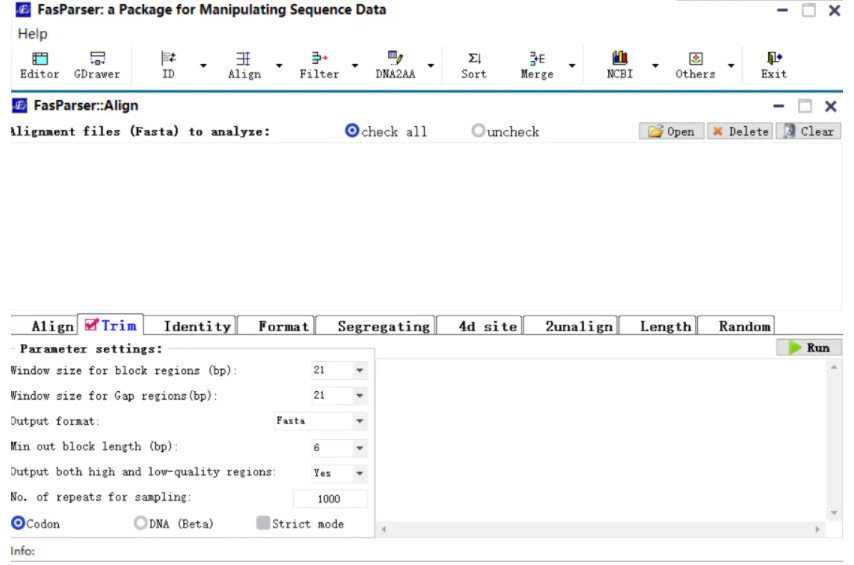
FasParser2该软件是由中国科学院昆明动物研究所孙艳波副研究员所开发 link地址,并于2018年在bioinformatics期刊发表。FasParser2主要是可以在友好界面窗口下对一些常规序列的操作,尤其是对多基因串联合并、序列提取、低质量序列鉴定、快速寻找趋同氨基酸变异位点等进行批处理化。尤其是,该软件增加了有效过滤序列比对质量的模块(滑窗分析),可实现序列中对非同源序列的清除,适用于大规模检测序列中经历达尔文正选择的位点,以及重建系统发育等分析。软件作者通过序列模拟测试发现,其修剪效率非常高,它能够删除95%以上的错误列,且基本不依赖于比对软件的选择,明显优于其他同类程序,如Guidance和Gblocks。另外,经过AlignTrim处理后的比对结果,可显著降低正选择检测中的检阳性事件(低于5%),且不会导致正选择的检测率降低,从而提高了检测正选择基因的准确性。该作者,将其与Gblocks、trimAl软件进行了比较,结果发现FasParser2的处理效果要优于这两款软件。
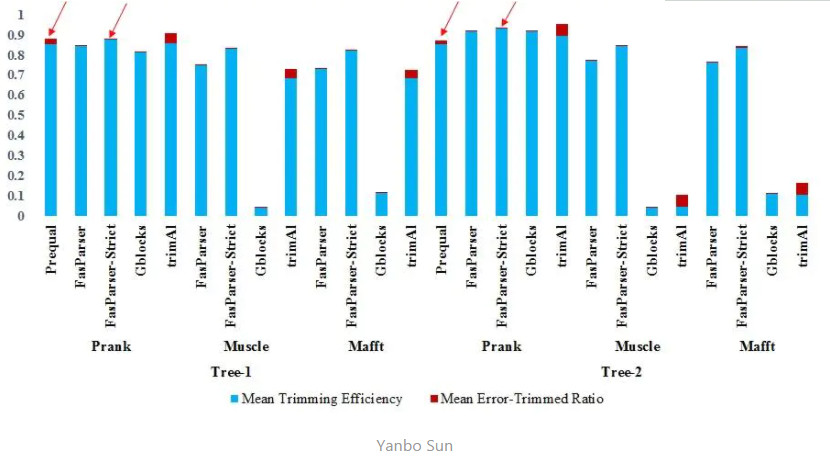
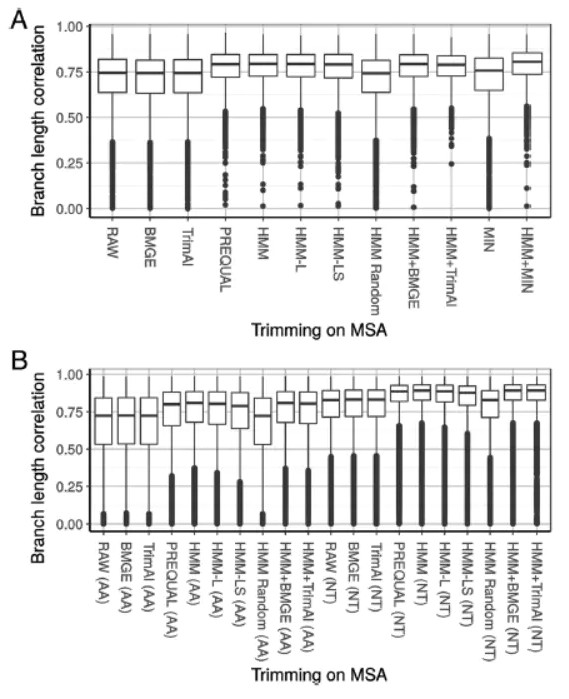
HmmCleaner与PREQUAL这两款软件也是近两年新发表的软件,它们采取了新的过滤算法(主要基于隐马尔可夫模型),能够准确识别多序列比对中的非同源片段,排除了由于测序质量、基因注释以及可变剪切造成的序列错误,其准确度以及速度相对较快,适用于大规模数据分析(Di Franco A. 2019;Whelan S. 2018)。值得注意的是,PREQUAL针对于非比对的蛋白编码序列以及氨基酸序列,而HmmCleaner只能操作氨基酸序列(可以是比对后的)。另外,它们对输入文件要求较高,一般fasta格式,序列中只能存在Gap和序列,不允许存在“?”、“!”、“X”等非编码字符,还要注意换行符问题。

03. Know Your Limits
- 数据类型: DNA vs. RNA, coding vs. non-coding nucleotides (wobble bp), AAs, proteins, etc.
- 数据特性:substitution (≠ mutation) rate strength (↑ vs. ↓), indel size and rate (% gap & gap length), pairwise sequence identity (PID), etc.
- 数据矩阵特性:# of tips, # of sequences, (alignment length ∝) data matrix weight, e.g., light (K, M) vs. heavy (G, T), etc.
- 计算资源:CPU time and RAM memory
04. Summary
每个人的数据可能与其他人的数据有很大差别,一定要学会正确选择适合自己数据的软件类型,这样得到的结果才是准确、可靠、可重复的。
生物信息学与计算机类似,更新速度很快,几年前还在使用的经典软件,现在可能已经过时了,因而保持与时俱进对生物信息人员是十分重要的。
最后,感谢中国科学院昆明动物所孙艳波老师给与的指导与帮助。
05. 这里放一张处理序列中存在移码突变的序列比对流程,已亲测(欢迎交流)


参考文献:
- Blackburne B P, Whelan S. Class of multiple sequence alignment algorithm affects genomic analysis[J]. Molecular biology and evolution, 2012, 30(3): 642-653.
- Löytynoja A. Phylogeny-aware alignment with PRANK[M]//Multiple sequence alignment methods. Humana Press, Totowa, NJ, 2014: 155-170.
- Löytynoja A, Goldman N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis[J]. Science, 2008, 320(5883): 1632-1635.
- Fletcher W, Yang Z. The effect of insertions, deletions, and alignment errors on the branch-site test of positive selection[J]. Molecular biology and evolution, 2010, 27(10): 2257-2267.
- Ranwez V, Douzery E J P, Cambon C, et al. MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codons[J]. Molecular biology and evolution, 2018, 35(10): 2582-2584.
- Sun Y B. FasParser2: a graphical platform for batch manipulation of tremendous amount of sequence data[J]. Bioinformatics, 2018, 34(14): 2493-2495.
- Whelan S, Irisarri I, Burki F. PREQUAL: detecting non-homologous characters in sets of unaligned homologous sequences[J]. Bioinformatics, 2018, 34(22): 3929-3930.
- Di Franco A, Poujol R, Baurain D, et al. Evaluating the usefulness of alignment filtering methods to reduce the impact of errors on evolutionary inferences[J]. BMC evolutionary biology, 2019, 19(1): 21.